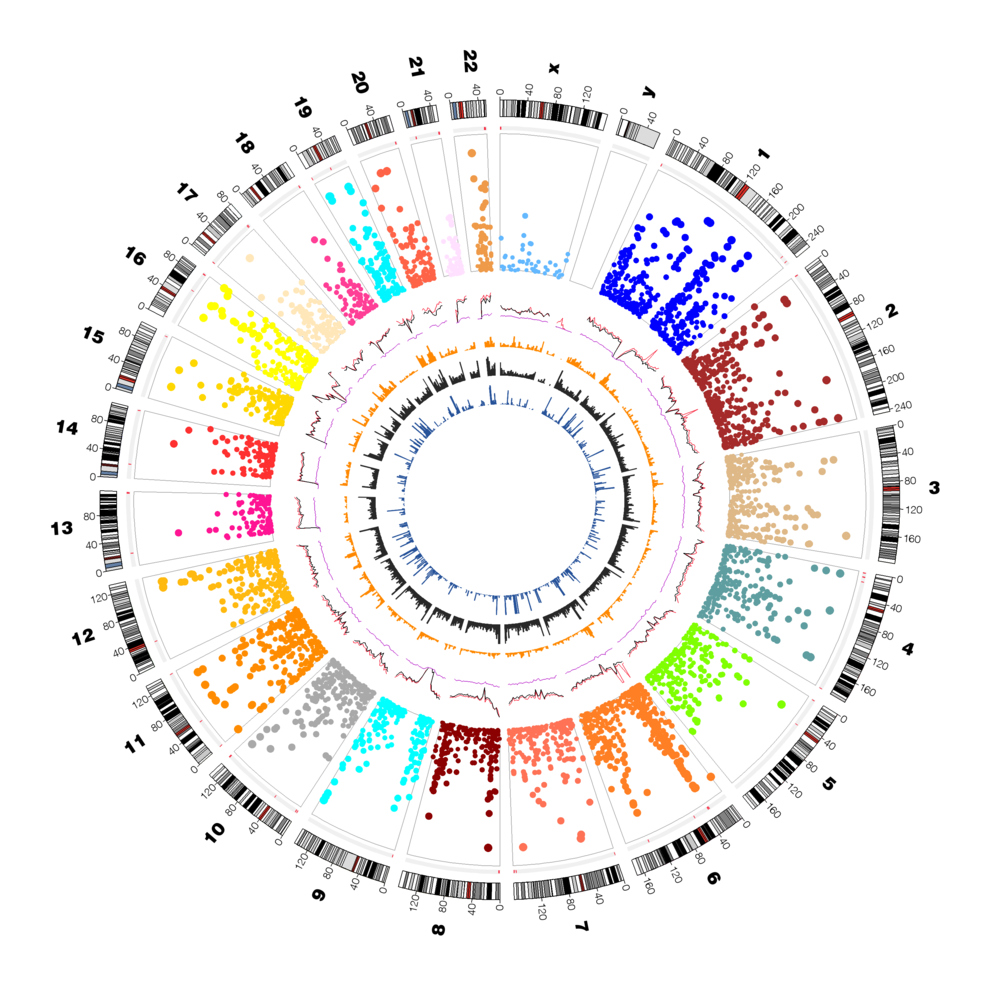

O GWAS nada mais é do que uma ferramenta utilizada no estudo de relação entre características fenotípicas e genótipo.
Afinal, todos já sabemos que exite um código por traz de cada característica expressa, em todos os organismos. Mas, o que você talvez não tenha entendido ainda, é sobre como é esclarecida a associação entre características individuais e gene (ou conjunto de genes associados).
Afinal, como essas características (fenótipo) são estudadas, ao ponto de se determinar que é relacionada a determinado conjunto de genes e não a outro conjunto, ou a um gene único específico? Fascinante, não?! É sobre isso que falaremos aqui.
O QUE É O GWAS?
O GWAS é uma metodologia usada para detectar associações entre variações genéticas e uma determinada características de interesse.
O estudo de associação genômica ampla ou Genome Wide Association Studies (GWAS) foi inicialmente desenvolvido para estudos epidemiológicos em humanos, mas já vem sendo muito utilizado em diferentes estudos animais e vegetais. Esse estudo busca associar regiões do genoma com características de interesse, para identificar possíveis regiões de maior efeito sobre um determinado fenótipo.

POR QUE OS ESTUDOS DE ASSOCIAÇÃO SÃO IMPORTANTES?
Uma das principais vantagens do GWAS é que a partir dele é possível testar um grande número de marcadores e várias características ao mesmo tempo. Como resultado disso, um grande número de regiões podem ser associadas.
Com essas informações, os melhoristas podem compreender melhor as características que estão estudando e identificar mais facilmente os genes envolvidos com essas características.
O GWAS E OS MARCADORES MOLECULARES
Sabemos que as variações genéticas entre indivíduos podem causar diferenças na expressão dos fenótipos. Mas como saber se as variações realmente possuem ligação com uma determinada característica?
Marcadores moleculares são a chave para o acesso a essas informações. Por meio deles, dados são gerados e coletados para descobrir as variações comuns em indivíduos com e sem a característica de interesse em todo o genoma.
Geralmente são utilizados SNPs (Simple Nucleotide Polymorphism). Se houver maior frequência nas variações nos indivíduos com a característica de interesse, dizemos que há uma associação. A partir daí, a utilização do GWAS é útil na validação da associação entre o SNP e a característica.
VANTAGENS DO MARCADOR SNP PARA A GWAS
É difícil identificar grandes associações com uma grande quantidade de dados genômicos. Assim, os marcadores moleculares auxiliam nesse processo.
Entre as classes de marcadores mais usadas atualmente estão os SNPs (Simple Nucleotide Polymorphism). São conhecidos como variações de nucleotídeos ou pequenas inserções/deleções que ocorrem em sequências de DNA homólogas.
Além disso, o GWAS requer uma alta densidade de marcadores, possível pela disponibilidade de tecnologias de genotipagem e sequenciamento. Os marcadores SNP são ótimas opções, pois são polimorfismos com alta densidade no genoma.
MECANISMO DO GWAS
O poder do GWAS em identificar uma verdadeira associação entre um SNP e uma característica vai depender da variância fenotípica dentro da população que foi explicada pelo marcador.
A variância fenotípica será determinada em razão de como as duas formas alélicas diferem em seu efeito fenotípico e sua frequência na amostra. A análise estatística é realizada para indicar a probabilidade de uma variante ser associada a uma característica.
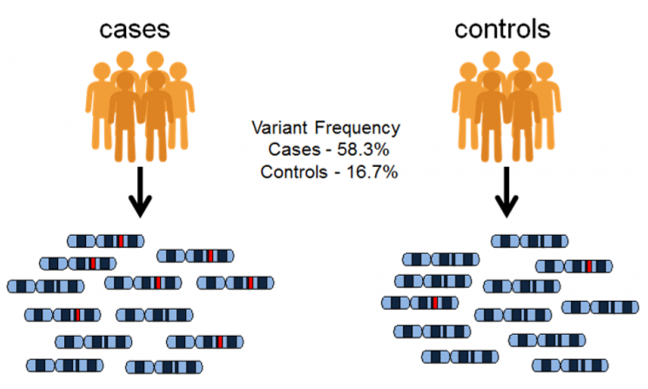
GWAS USA O PADRÃO DE DESEQUILÍBRIO DE LIGAÇÃO (LD)
O GWAS usa o padrão de desequilíbrio de ligação (LD) em uma população para mapear regiões.
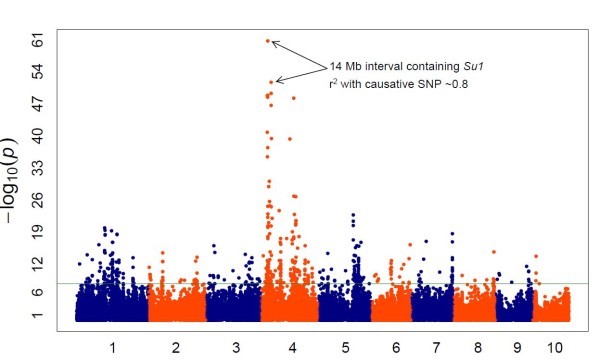
Isso ocorre porque marcadores moleculares vizinhos tendem a ser herdados juntos, estando assim em desequilíbrio de ligação (LD) entre os dois marcadores. Ao final, podemos observar os marcadores SNP mais interessantes por meio de um Gráfico de Manhattan. Os pontos mais altos correspondem a regiões genômicas com uma forte associação com a característica estudada.
GWAS NA SELEÇÃO ASSISTIDA POR MARCADORES E PROGRAMAS DE MELHORAMENTO
A partir do GWAS é possível detectar e identificar regiões importantes, loci de características quantitativas (QTLs) e genes que controlam e explicam determinados fenótipos em cultivares e em acessos de germoplasma.
Assim, o objetivo é obter o máximo de proveito dessa exploração da diversidade genética e identificar novas combinações alélicas de genes relacionados a caracteres de importância agronômica.
Essas informações contribuem significativamente para a obtenção de cultivares mais produtivas, tolerantes ou resistentes à determinada doença ou parasita. Assim, o GWAS pode ser incorporado na seleção assistida por marcadores (SAM) e auxiliar programas de melhoramento.
GWAS E O SUCESSO EM CULTURAS
O GWAS foi conduzido e com sucesso em diversas culturas de cereais como trigo (Arora et al. 2019), arroz ( Quero et al. 2018 ), sorgo ( Morris et al. 2013 ), milho ( Xiao et al. 2017 ), soja (Zhang, 2018), eucalipto (Müller, 2018), arvores frutíferas (Iwata, 2016) para seleção de várias características de importância econômica.
Fontes
Yang, J. et al. Genome-wide complex trait analysis (GCTA): methods, data analyses, and interpretations. In: Genome-wide association studies and genomic prediction. Humana Press, Totowa, 2013.
Zhang J. et al. Genome wide association study, genomic prediction and marker assisted selection for seed weight in soybean (Glycine max). Theor Appl Genet, 2016.
Romay, M. C. et al. Comprehensive genotyping of the USA national maize inbred seed bank. Genome biology, 2013.
Arora, S. et al. Genome-wide association mapping of grain micronutrients concentration in Aegilops tauschii. Frontiers in plant science, 2019.
Müller, B. S. et al. Independent and Joint‐GWAS for growth traits in Eucalyptus by assembling genome‐wide data for 3373 individuals across four breeding populations. New Phytologist, 2019.
Iwata, H. et al. Genomics-assisted breeding in fruit trees. Breeding science, 2016.
Zhang, J. et al. Genome-wide scan for seed composition provides insights into soybean quality improvement and the impacts of domestication and breeding. Molecular plant, 2018.
Quero, G. et al. Genome-wide association study using historical breeding populations discovers genomic regions involved in high-quality rice. The plant genome, 2018.
Morris, G. P. et al. Dissecting genome-wide association signals for loss-of-function phenotypes in sorghum flavonoid pigmentation traits. G3: Genes, Genomes, Genetics, 2013.